
Charakterystyka sulfidów
Wiadomości ogólneSulfidy (tioetery) to związki zawierające dwuwartościowy atom siarki i które możemy zapisać ogólnym wzorem RSR, gdzie R może być grupą alkilową, winylową, allilową, arylową itp. Merkaptale – R1R2C(SR3)2 oraz ortotioestry – RC(SR’)3 także możemy zaliczyć do tej grupy związków.
Na przestrzeni lat chemia sulfidów bardzo mocno rozwinęła się i grupa tych związków jest coraz lepiej poznana. Związki te budzą takie zainteresowanie ze względu na łatwość z jaką możemy je otrzymać z nieorganicznych sulfidów które dość licznie występują w przyrodzie. Wiele sulfidów można wyizolować z roślin. Sulfid metylowy występuje w sokach wielu roślin w tym w geranium i mięcie. Odpowiada również za specyficzny zapach eukaliptusa. Sulfid allilowy jest odpowiedzialny za charakterystyczny zapach czosnku. Inne sulfidy również występują w roślinach, np. metylmerkaptopropanol został wyodrębniony z oleju sojowego. Sulfidy występują również w allium ursinum oraz allium sativum. Rośliny te zawdzięczają swoje antybakteryjne właściwości obecnością C3H5SO*SC3H5. Sulfidy są także substratami licznych reakcji otrzymywania innych związków zawierających siarkę takich jak: sulfoksydy, sulfony itd. [7].
Otrzymywanie sulfidówWiększość metod otrzymywania sulfidów została opracowana jeszcze w latach sześćdziesiątych ubiegłego stulecia. W późniejszym czasie metody te zostały udoskonalone i rozwijane oraz wykorzystywane do otrzymywania także innych związków organicznych zawierających siarkę.
Alkilowanie tioli
Alkilowanie tioli
Metoda ta jest najpopularniejszą laboratoryjną metodą otrzymywania sulfidów. Przebiega dość łatwo z zadowalającą wydajnością. Przebieg tej reakcji możemy ogólnie zapisać wzorem:
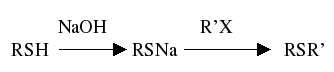
R, R’= alkil, aryl itp.
Addycja
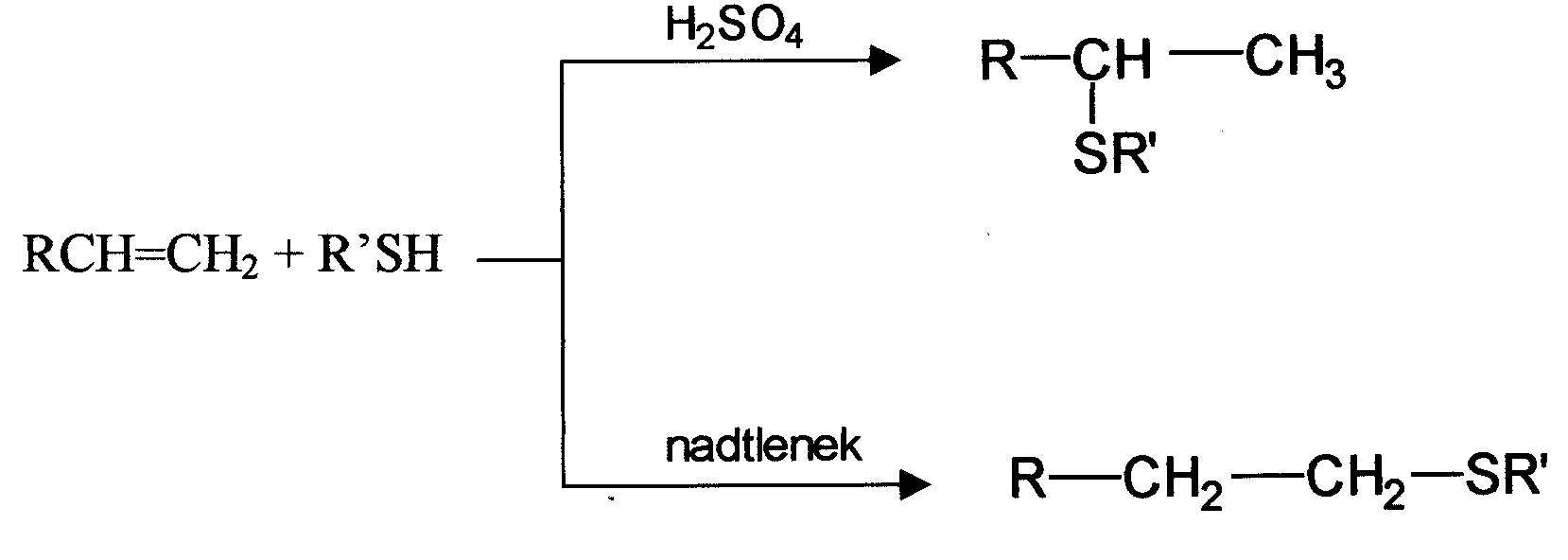
Reakcja addycji do wiązania wielokrotnego znalazła zastosowanie do syntezy różnych sulfidów. Przebiega ona według dwóch mechanizmów: reakcji jonowej oraz reakcji wolnorodnikowej. Na to jaki mechanizm będzie dominował podczas reakcji addycji wpływają różne czynniki takie jak: rodzaj reagentów, katalizator, itp. Reakcja jonowa katalizowana jest przez kwasy lub zasady i odbywa się zgodnie z reguła Markownikowa. Reakcja wolnorodnikowa jest inicjowana w obecności nadtlenków, azonitryli lub przez naświetlanie i wtedy przebiega niezgodnie z regułą Markownikowa. Schematycznie reakcje te możemy zapisać:
Cykloaddycja
Jest to fotochemiczna reakcja tioketonów z olefinami w tym również z dienami według mechanizmu addycji Dielsa-Aldera. Przebiega ona z dość dobrą wydajnością. Oto przykładowa reakcja dienu z tioketonem:
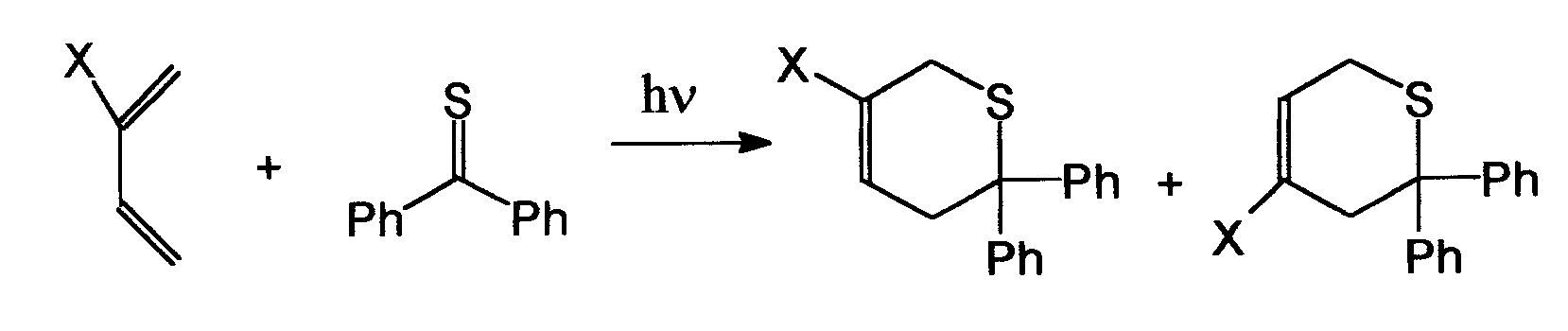
Rozkład disulfidów
Organiczne disulfidy nie są typowymi substratami wykorzystywanymi w tej reakcji aczkolwiek są one często stosowane. Rozpad wiązania S-S w disulfidzie odbywa się w reakcji z karboanionem, wolnym rodnikiem lub przez odszczepienie jednego atomu siarki przez odpowiednie związki. Oto przykłady niektórych takich reakcji [7]:
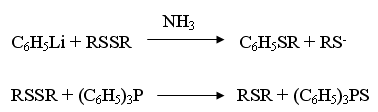
Właściwości fizyczne i chemiczne sulfidów
Dla wielu sulfidów określono energię dysocjacji wiązań aczkolwiek te wartości nie są tak pewne jak w przypadku analogicznych związków zawierających tlen. Energia wiązania C-S i S-H jest mniejsza niż w przypadku wiązania O-H i C-O. Długość wiązania C-S i C-O dla prostych sulfidów alkilowych i eterów jest podobna i wynosi odpowiednio 1,82 Å oraz 1,41 Å. Kąty między wiązaniami C-S-C a C-O-C wynoszą odpowiednio 105º i 112º. W większości przypadków wiązania siarki są dłuższe a kąty mniejsze niż w wypadku analogicznych związków zawierających tlen [8]. Oto kilka przykładów:
| Związek | Długość wiązania C-S lub C-O [Å] | Kąt między C-S-C lub C-O-C [º] |
| (CH3)2S | 1,82 | 105 |
| (CF3)2S | 1,83 | 105,6 |
| (p-CH3C6H4)2S | 1,75 | 109 |
| (p-BrC6H4)2S | 1,75 | 109 |
| (C6H5)2S | 113 | |
| (CH3)2O | 1,41 | 111,7 |
Zdarzają się jednak wyjątki od tej reguły spowodowane obecnością specyficznych podstawników czy strukturą cząsteczki (np. wielkością i kształtem pierścienia cyklicznego). Przykładem takiego związku jest sulfid arylowy, który ma krótsze wiązanie C-S i większy kąt między wiązaniami C-S-C niż sulfidy alkilowe. Różnicę w tych wartościach powodują elektrony π pierścienia aromatycznego.
Sulfidy przejawiają również właściwości donorowe. Zasadowość albo moc donorowa atomów siarki w sulfidach jest jedną z ważniejszych właściwości tej grupy związków i jest ona blisko związana z reaktywnością sulfidów. Zasadowość tę możemy lepiej wytłumaczyć jeśli przypatrzymy się bliżej równowadze kwas–zasada i budowie kompleksów charge-transfer. Dokładniejszych informacji może nam dostarczyć analiza widm NMR.
Sulfidy jako zasady Lewisa mogą wytwarzać wiązanie wodorowe z kwasami. W tym przypadku wiązanie takie jest dość słabe, a jego tworzenie może być dodatkowo osłabiane jeśli związek zawiera więcej niż jedno miejsce donorowe. Przykładem może być tworzenie się wiązania wodorowego między różnymi fenolami i sulfidami [9]:
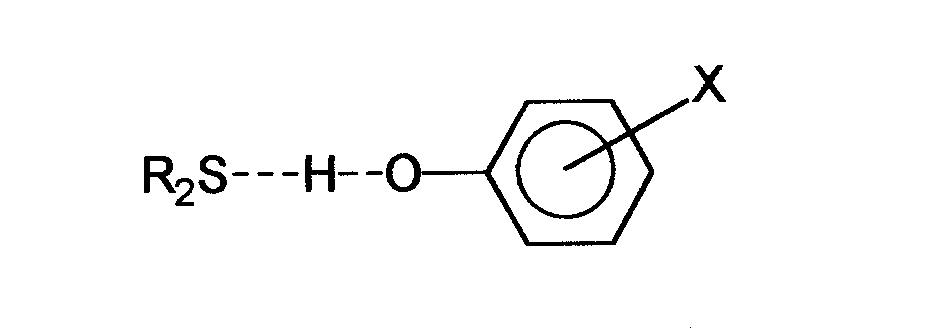
Sulfidy mogą tworzyć również kompleksy z metalami wytwarzając wiązania siarka-metal o różnej sile w zależności od rodzaju metalu. Do tej pory poznano ogromną liczbę takich kompleksów. Wiązanie tworzące się w kompleksach z metalami alkalicznymi jest generalnie słabe, podczas gdy wiązanie z metalami ciężkimi takimi jak rtęć czy miedź jest silne i czasem utworzenie takiego wiązania może spowodować rozerwanie się wiązania C-S. Halogenki metali III grupy tworzą wiązania o średniej sile. Alifatyczne sulfidy tworzą w tym wypadku dość silne kompleksy w przeciwieństwie do sulfidów aromatycznych, które tworzą dość słabe wiązania. Obecnością wolnych par elektronowych atomu siarki sprzęgających się z elektronami pierścienia aromatycznego powoduje, że elektrony te tworzą wiązania S-metal dużo słabsze niż w przypadku sulfidów aromatycznych. Znane są również kompleksy sulfidów z halogenkami [10].
Reakcje sulfidów
1. Reakcja addycji
Pierwszym typem reakcji jakim ulegają sulfidy jest addycja halogenków i utworzenie odpowiednich halogenopochodnych. Najłatwiej ulegają tej reakcji sulfidy alkilowe, nieco słabiej arylowe z tym, że podczas addycji może nastąpić migracja atomu halogenka do pierścienia aromatycznego.
Kolejnym typem reakcji jakim, ulegają sulfidy jest addycja soli metali ciężkich i tworzenie odpowiednich kompleksów. Najczęściej spotykanymi kompleksami są kompleksy z solami rtęci, platyny, palladu, złota oraz srebra. Pierwsze próby takiej syntezy przeprowadził Mabery, który na wyizolowane z wysokosiarkowej ropy naftowej sulfidy podziałał alkoholowym roztworem chlorku srebra. W wyniku tej reakcji wytworzyło się kilka kompleksów w których stosunek sulfidu do soli rtęci wynosił 1:2, 1:1, 2:1 itp. Kompleksy te mogą przekształcać się wzajemnie w siebie tak, że zawartość poszczególnych kompleksów w roztworze może być różna. Aby otrzymać w miarę jednolity produkt stosuję się rekrystalizację z różnych rozpuszczalników. Przykładem może być kompleks chlorku rtęci oraz sulfidu propylowego gdzie w alkoholowym roztworze dominuje kompleks Pr2SּHgCl2 który w roztworze benzenu rekrystalizuje w Pr2Sּ2HgCl2 [7]. Do najczęściej spotykanych kompleksów jakie tworzą sulfidy z solami platyny należą: PtX2ּ2R2S oraz PtX4ּ2R2S, które tworzą izomery cis i trans. Mogą powstawać również znacznie bardziej skomplikowane kompleksy jak np.: PtX2X’2ּ2R2S.
2. Reakcja rozkładu
Reakcja ta najczęściej polega na rozerwaniu wiązania węgiel-siarka. Jako pierwsi reakcję tego typu opisali Tarbell i Harnish [11]. Polegała ona na katalitycznym krakingu odpowiednich sulfidów w wysokich temperaturach. W temperaturze 230˚C cząsteczki sulfidu benzylowego rozpadały się w 95%, allilowego w 61%, a propylowego w 21%. Produktem tego rozkładu były między innymi małe cząsteczki merkaptanów, tiofenów, olefin i parafin [7].
Wysokociśnieniowa reakcja krakingu z zastosowaniem wodoru prowadzi do utworzenia się cząsteczek merkaptanu odpowiedniego węglowodoru, a przeprowadzana w obecności katalizatorów niklowych, kobaltowo-molibdenowych prowadzi do powstania siarkowodoru oraz odpowiednich węglowodorów [9]. Rozerwanie wiązania węgiel-siarka następuje również w reakcji z solami Grignarda orzaz z BrCN.
3. Reakcje z metalami
Jednym z przykładów tego typu reakcji jest reakcja sulfidu metylowoetylowego z BuLi, w efekcie której lit przyłącza się do grupy metylowej sulfidu i może ulegać dalszym reakcjom jak np. z dwutlenkiem węgla. Gdy zamiast grupy metylowej w sulfidzie występuje inna, dużo większa grupa to wtedy preferowane jest przyłączenie się atomu litu do pierścienia aromatycznego. Jeśli na sulfid metylowoetylowy podziałamy octanem rtęci to w tym wypadku najbardziej preferowane będzie przyłączenie się atomu rtęci razem z grupą octanową do pierścienia aromatycznego.
4. Reakcja tworzenia się soli sulfoniowych
Kolejną ważną reakcją jakiej ulegają sulfidy alifatyczne jest ich łączenie się z halogenkami alkilowymi z utworzeniem związków sulfoniowych. Są to sole dobrze rozpuszczalne w wodzie, które mają podobną budowę i właściwości do soli tetraalkiloamonu. Pierwsze związki sulfoniowe powstały przez ogrzewanie sulfidu etylowego i jodku metylu:
MeI + Et2S  MeEt2SI
MeEt2SI
Szybkość tej reakcji zależy od wielkości wprowadzanej grupy. Jodek metylu reaguje już w temperaturze pokojowej i szybkość tej reakcji jest ponad osiemdziesięciosiedmiokrotnie większa niż w przypadku jodku etylu. Może on również reagować z wyższymi sulfidami alkilowymi i mieszanymi sulfidami takimi jak: metylocykloheksylo-, metyloamylo-, dimetoksymetylo-, benzylo sulfidami. Jodek trietylosulfoniowy ma kształt rombu. Bardzo dobrze rozpuszcza się w wodzie i występuje w niej w postaci jonów. Jon R3S+ ma kształt piramidy z atomem siarki na szczycie.[7, 9]
Zastosowanie sulfidów
Niektóre sulfidy i ich mieszaniny z innymi związkami wykorzystywane są w środkach owadobójczych i grzybobójczych. Wiele sulfidów aromatycznych takich jak: sulfid nitrofenylowy, nitrotienylowy charakteryzuje się właściwościami bakteriobójczymi i znajduje zastosowanie preparatach dezynfekujących. Disulfid PhSCH2CH2SPh jest dość silnie toksyczny i wykorzystuje się go do niszczenia larw komarów. Sulfid metylowy, merkaptan etylu są silnymi związkami wabiącymi różne owady np. muchy mięsne. Niektóre sulfidy są użyteczne w procesie wulkanizacji, inne są składnikami mydeł i kremów. Wykorzystuje się je również jako składnik polimerów kauczukopodobnych i żywic. Etylofenylosulfid stabilizuje żywicę polisulfonową. Selenki sulfidów alkilowych znajdują zastosowanie jako bardzo efektywne antyutleniacze w olejach naturalnych. Sulfidy znajdują zastosowanie również w innych gałęziach przemysłu, w wielu procesach technologicznych [9].
Mimo, że siarka stanowi w organizmach żywych stanowi zaledwie 2% wszystkich pierwiastków to jednak trudno przecenić rolę jaką odgrywają związki siarki. Reakcje redox między tiolami i disulfidami są odpowiedzialne za stabilizację przestrzennej struktury białek oraz za ich oddziaływanie z enzymami. Stanowią również część koenzymu, kwasu liponowego oraz innych koenzymów, odgrywając zasadniczą rolę w ich działaniu między innymi podczas syntezy kwasu tłuszczowego [12].
komentarze
Copyright © 2008-2010 EPrace oraz autorzy prac.